2899/AB XXI.GP
Eingelangt am: 07.12.2001
Bundesministerium für soziale Sicherheit und
Generationen
Ich beantworte die an mich gerichtete
schriftliche parlamentarische Anfrage der
Abgeordneten
Mag. Johann Maier und GenossInnen, Nr. 2921, wie folgt:
Fragen 1, 2 und 3:
Mein Ministerium wurde erstmals im
Juni 2001 von Großbritannien (= EU-
Referenzstaat
für das im gegenseitigen Anerkennungsverfahren in der EU
zugelassene Lipobay) und der Fa. Bayer informiert, dass eine Fach- und
Gebrauchsinformationsänderung für Lipobay erforderlich ist (Aufnahme
eines
Hinweises über vermehrtes Auftreten von Rhabdomyolyse bei gleichzeitiger
Anwendung mit Gemfibrozil /EU-Dringlichkeitsverfahren: Dauer 21. - 29. Juni
2001).
Ein diesbezüglicher Erlass meines Ressorts erfolgte am 29. Juni 2001. Am
7. bzw. 8.
August 2001 teilten
Großbritannien und die Fa. Bayer mit, dass Bayer Lipobay
freiwillig vom Markt nehmen wird (Begründung der Fa. Bayer:
ungenügende
Beachtung der Warnhinweise vom Juni 2001). Am 9. August 2001 erfolgte der
Erlass
meines Ressorts zur
Marktrücknahme.
Fragen 4 und 5:
Der erste Verdachtsfall einer UAW
(unerwünschten Arzneimittelwirkung) mit Lipobay
wurde meinem Ressort am 25. Februar 1999 gemeldet. Diese Meldung wurde
umgehend der Firma Bayer mitgeteilt (gesetzliche Verpflichtung gemäß
§ 75 des
Arzneimittelgesetzes
i.d.g.F.). Gleichzeitig wurde die Meldung - wie jede UAW-
Meldung - einer
pharmakologischen Begutachtung unterzogen. Eine Maßnahme
wurde damals aus fachlicher Sicht auf Grund dieses einen Falles noch nicht
für
erforderlich
gehalten.
Frage 6:
Bis 30. September 2001 sind in
meinem Ressort 8 UAW-Meldungen im
Zusammenhang mit der
Anwendung von Lipobay eingelangt.
Frage 7:
In diesem
Zusammenhang wäre unter Umständen an Maßnahmen auf dem
Zivilrechtsweg zu denken. Die Beurteilung der diesbezüglichen
Möglichkeiten obliegt
jedoch nicht meinem Ressort, sondern dem Bundesministerium für Justiz.
Frage 8:
Mein Ministerium
erhält laufend UAW-Meldungen aus Österreich. Jährlich werden
seitens der Ärzteschaft an die tausend UAW-Fälle gemeldet. Jede einzelne
Meldung
wird umgehend dem betroffenen Zulassungsinhaber mitgeteilt und pharmakologisch
bewertet. Alle Aufhebungen von Zulassungen erfolgten nicht ausschließlich
auf
Grund von österreichischen Meldungen. Diese Entscheidungen werden immer
unter
Zugrundelegung des Arzneimittelinformationssystems der EU getroffen.
Fragen 9 und
13:
In der EU gibt es
bereits seit Jahren ein Arzneimittelinformationssystem, in das jeder
EU-Staat miteingebunden ist. Der laufende Informationsaustausch erfolgt in Form
von Fax und/oder e-mail-Mitteilungen. Es gibt für nicht dringende
Fälle das NUI (Non
Urgent
Information-System, in dringenden Fällen wird das RA (Rapid Alert)-System
in
Anspruch genommen.
Frage 10:
Ja, die Anforderungen
an ein Zulassungsdossier sind strikt geregelt und werden
laufend dem neuesten Wissensstand angepasst. Das europäische
Zulassungssystem beruht in seinen wesentlichen Grundsätzen auf dem Konzept
der
Zusammenführung wissenschaftlichen Sachverstandes. Dies gilt im besonderen
natürlich für das zentrale Zulassungsverfahren, bei dem die Bewertung
durch ein aus
Sachverständigen der einzelnen Mitgliedstaaten zusammengesetztes
Expertengremium im Rahmen der Europäischen Arzneimittelagentur erfolgt,
hat aber
natürlich auch für das dezentrale Verfahren Gültigkeit, wo im
“gemeinschaftlichen"
Verfahrensteil eine Abklärung der von den einzelnen Mitgliedstaaten
geäußerten
Punkte bzw. Bedenken auf europäischer Ebene stattfindet.
Im Rahmen einer Bewertung dieser
Zulassungsverfahren im Sinne einer Review der
pharmazeutischen Gesetzgebung der EU werden Überlegungen im Zusammenhang
mit der Steigerung der Leistungsfähigkeit der einzelnen Verfahrensmodule
natürlich
auch im Hinblick auf die für die
Erteilung von Zulassungen entscheidenden
Parameter Qualität, Sicherheit und Wirksamkeit angestellt.
Im Übrigen sind die Anforderungen, die in der EU an die
Zulassungsunterlagen
gestellt werden, mit jenen in den USA bzw. Japan vergleichbar.
Frage 11:
Österreich ist entsprechend
seiner Verpflichtungen aus der EU-Mitgliedschaft in das
gemeinschaftliche
Arzneimittelsystem der EU eingebunden.
Frage 12:
Eine dringende
Mitteilung zur Arzneimittelsicherheit wird an die Angehörigen der
Sanitätsberufe (Ärzte, Apotheker etc.) mittels Erlass an die
Landessanitätsdirektoren,
Ärzte-, Apothekerkammer etc. vorgenommen.
Frage 14:
Transparenz der
Entscheidungsgrundlage für Zulassungen ist wichtig und man ist
seitens der Zulassungsbehörden bemüht diese ständig zu erhöhen.
Derzeit werden
EPAR's (European Public Assessment Reports) von der Agentur in London zu
zentralen Zulassungen auf Basis der Bewertungsberichte von Rapporteur und Co-
Rapporteur im Internet veröffentlicht. Da alle neuen Wirkstoffe nahezu
ausschließlich
im zentralen Verfahren zugelassen werden, ist dies hier besonders wichtig. Die
Veröffentlichung von PAR's ist derzeit auch für Präparate, die
im gegenseitigen
Anerkennungsverfahren zugelassen werden in Planung, damit wären dann alle
neuen Substanzen abgedeckt. In weiterer Folge wird diskutiert PAR's auch
für
Produkte, die im rein nationalen Verfahren zugelassen werden, zu
veröffentlichen.
Es ist diese Steigerung der Transparenz sicher wünschenswert, jedoch ist
diese
zusätzliche Aufgabe nur zu bewältigen, wenn den nationalen
Behörden
diesbezüglich mehr Personal zur Verfügung gestellt wird.
Fragen 15
und 16:
Generell kann man
sagen, dass die Behörde bestrebt ist, die gesetzlich geregelten
Fristen einzuhalten und die Dauer der Zulassungsverfahren mit Hilfe von
elektronischen Datensysternen besser zu dokumentieren. Dies bedeutet, dass
früher
das Datum der Einreichung des Zulassungsantrages sowie das Zulassungsdatum
für
die “Dauer des Zulassungsverfahrens" erfasst wurden. Heute wird
dahingehend
differenziert, ob die Zeit zu Lasten der Behörde (für
Bewertungstätigkeit) oder der
antragstellenden Firma (bei aufgezeigten Mängeln) geht. Dieses System
findet bei
zentralen Zulassungen bereits Anwendung, für nationale Anträge werden
derzeit
bestehende
Datenbanksysteme überarbeitet.
Eine Änderung in der Dauer
der Zulassungsverfahren hat sich dadurch ergeben,
dass es seit 1995
neben dem rein nationalen Zulassungsverfahren noch das
zentrale Verfahren
und das gegenseitige Anerkennungsverfahren gibt.
Frage 17:
Entsprechend einer
diesbezüglichen Vorgabe in den europäischen Richtlinien
betreffend Arzneimittel wurde in der dem Parlament vorliegenden Novelle zum
Arzneimittelgesetz die Frist für Entscheidungen in einem
Zulassungsverfahren auf
sieben Monate
festgelegt.
Frage 18:
Zentrale Zulassungen
gemäß Verordnung 2309/93 gibt es erst seit 1995,
Inkrafttreten des zentralen Zulassungsverfahrens mit 1.1.1995, seit diesem
Zeitpunkt
sind für 190 Produktgrupper die EU-Zulassungen ausgesprochen worden.
Fragen 19 und
20:
Es wurden keine
Zulassungen nach §11 Abs. 1 Z2 sowie § 11 Abs. 1 Z3
ausgesprochen.
Frage 21:
Zulassungsverfahren: rein national und MRP
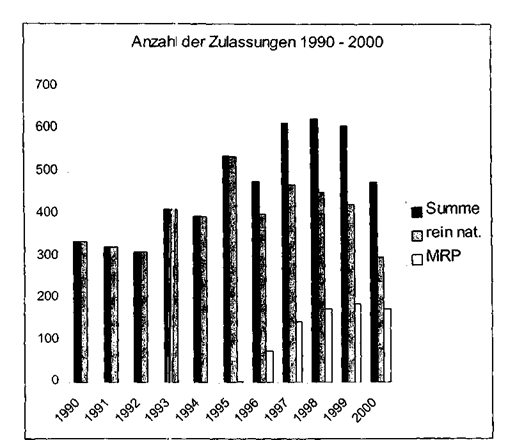
zusätzlich
gelten auch die EU-Zulassungen für den österreichischen Markt.
Frage 22:
Von Amtswegen wurden in den Jahren 1990 -
1998 keine Zulassungen aufgehoben.
1999 waren es 4 Präparate
2000 waren es 30 Präparate
von 1.1.2001
bis 30.9.2001 ist es 1 Produkt.
Frage 23:
Auf Grund der Erfüllung der
Voraussetzung nach § 72 Abs. 1 Z2 AMG haben sich
von 1990 bis 30.9.2001 2362
Personen für den Beruf des Pharmareferenten
qualifiziert.
Die Aufschlüsselung lautet:
1990 : 199
1991
: 198
1992
: 230
1993
: 265
1994
: 203
1995
: 176
1996
: 214
1997
:
191
1998
:
196
1999
:
192
2000
:
168
2001 (bis 30.9.): 130
Frage 24:
Für die Ausübung des
Pharmareferentenberufes besteht keine Meldepflicht, daher
ist mir auch nicht bekannt, wie viele Pharmareferenten derzeit aktiv sind.
Pharmareferenten sind gegenüber
ihrem Auftraggeber verantwortlich. Die zentrale
Erfassung von Pharmareferentlnnen in einer eigenen Liste könnte die
Aufgabe einer
freiwilligen beruflichen Interessensvertretung der Pharmareferentlnnen sein.