14750/J XXVII. GP
Eingelangt am 30.03.2023
Dieser Text wurde elektronisch übermittelt. Abweichungen vom Original sind
möglich.
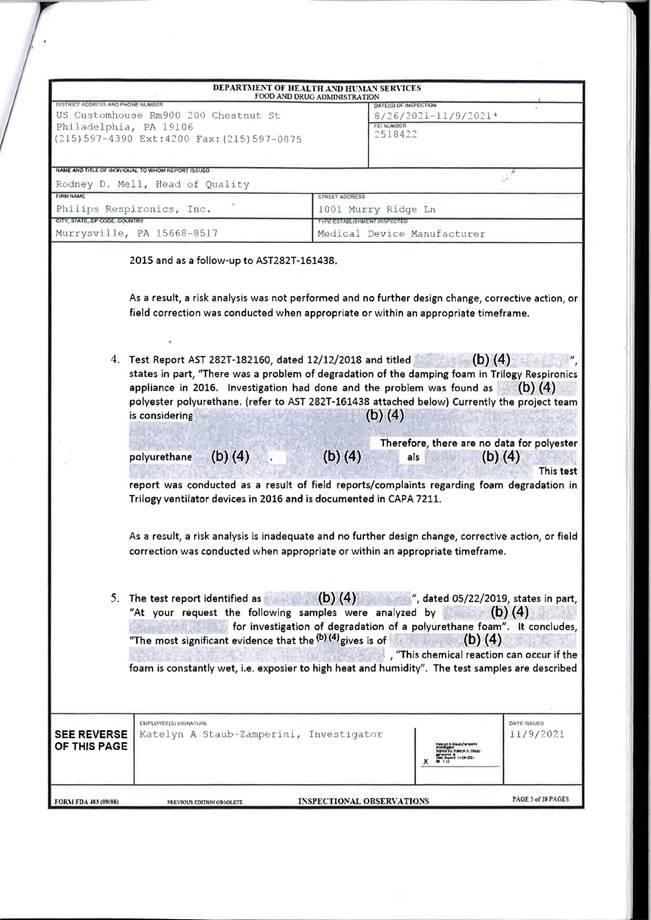
Anfrage
der Abgeordneten Philip Kucher, Mag. Christian Drobits,
Genossinnen und Genossen
an den Bundesminister für Soziales, Gesundheit, Pflege und Konsumentenschutz
betreffend Schadhafte Philips Respironics CPAP und Bi-Level PAP Beatmungs-Geräte trotz CE-Zertifizierung
Im Juni 2021 hat Philips Respironics eine „Dringende Sicherheitsmitteilung“ herausgegeben, die von der österreichischen Generalvertretung an alle Patienten geschickt wurde. Darin wird die mögliche „Anfälligkeit für Zersetzung des schalldämmenden Schaumstoffs (PE-PUR) und Freisetzung flüchtiger organischer Verbindungen sog. VOC´s“ angeführt Die Schaumstoff-Partikel können in die Atemwege von Patienten gelangen, und die Chemikalien können toxisch und krebserregend sein. Deshalb wurde vor möglichen Gesundheitsschäden wie u.a. Atemwegsproblemen, Entzündungen, Organschäden in Leber und Niere, Kopfschmerzen. Übelkeit u.v.a. gewarnt.
Ca. 35.000 Patienten sind in Österreich betroffen, da sie eines der in der Warnung angeführten CE zertifizierten Beatmungs-Geräte verwenden. Diese CE Zertifizierung bietet also keinerlei Sicherheit. Die Patienten sollten mit ihrem Lungenfacharzt eine individuelle Risikoabwägung vornehmen, ob der Schaden durch Verwendung oder Nicht-Verwendung größer wäre und wurden ersucht, bis zum möglichen Tausch ihres Gerätes zu warten. Es wurde in ärztlichen Stellungnahmen vor einem Absetzen der Beatmungstherapie gewarnt, jedoch konnten Patienten nicht wissen, ob ihr Gerät schadhaft ist. Dies führte zu einer enormen Verunsicherung in der Patientenschaft, da eine solche Abwägung nur auf „Annahmen“ und nicht auf Fakten beruhen konnte, und ihnen gleichsam nur die Möglichkeit bot, zwischen Pest oder Cholera zu wählen.
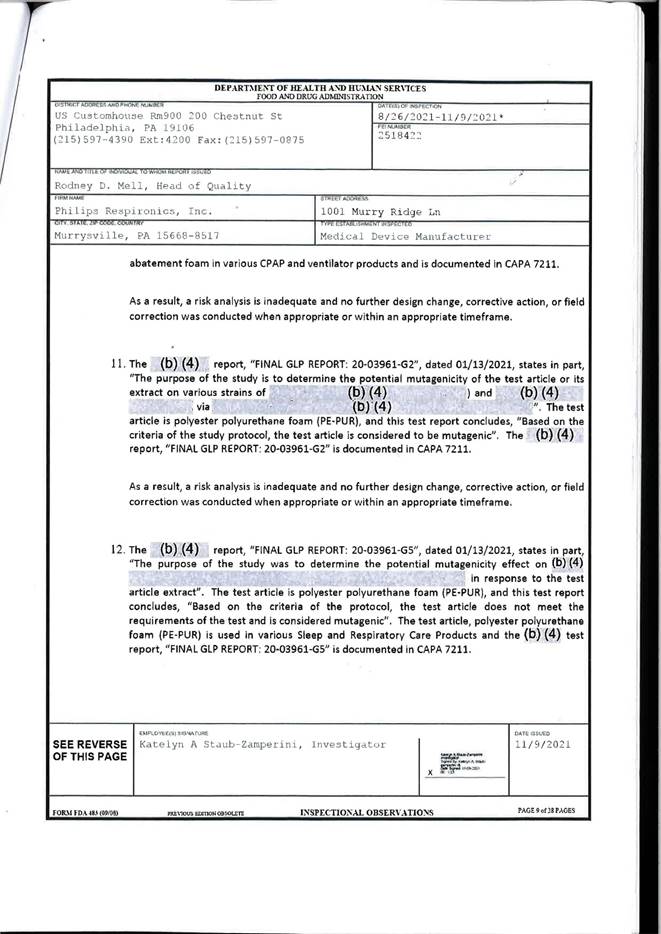
Entgegen zahlreicher Ankündigungen von Philips ist dieser Tausch ausgesprochen schleppend vor sich gegangen, zuerst war seitens Philips in einer ORF-Sendung vom Start im September 21 die Rede, tatsächlich wurde die ersten Geräte erst im Februar 2022 getauscht. Erschwerend kommt hinzu, dass die Food & Drug Administration in den USA einen Bericht herausgegeben hat, wonach die Herstellerin der fehlerhaften Beatmungsgeräte bereits im Jahr 2015 über die Zersetzung des Schaumstoffs in den Beatmungsgeräten gewusst haben soll. Dieser Bericht der FDA wird dieser Anfrage in beglaubigter Übersetzung beigelegt.
Seit April 2021 sind in den USA mehr 98.000 medical device reports bei der FDA eingegangen. Die auf den europäischen Binnenmarkt importierten Beatmungsgeräte sind baugleich mit den in den USA hergestellten dort verkauften Beatmungsgeräten. In diesen medical device reports wird von gravierenden Erkrankungen wie Krebs, Asthma u.a. berichtet.[1]
AUSTAUSCH
Laut der 11728/AB XXVII. GP – Anfragebeantwortung sollte der Austausch der Beatmungsgeräte mit dem 1.Quartal 2023 abgeschlossen sein. Laut dem aktuellen Philips update vom 19.3.2023 [2] wurden bisher 23 425 reparierte Geräte nach Österreich ausgeliefert, 35.000 sind in Österreich betroffen.
Die unterzeichneten Abgeordneten stellen daher folgende
Anfrage
1. Ist mit Ende März 2023 tatsächlich für alle Patienten der Gerätetausch abgeschlossen?
Qualitätskontrolle beim Austausch:
2. Wurden die Austauschgeräte von den öst. Firmen der Generalvertretung auf Schadhaftigkeit untersucht, speziell der Schaumstoff– wenn ja in welcher Art und Weise?
3. Welche Sicherheit haben betroffene Patienten, dass anschließend im Schadensfall tatsächlich eine Meldung an das BASG erfolgt ist?
4. Werden die schadhaften Geräte einer weiteren genaueren Analyse unterzogen, um für die Therapie der betroffenen Patienten wichtige Erkenntnisse zu gewinnen?
5. Werden die schadhaften Geräte für weitere Untersuchungen aufbewahrt?
MELDUNGEN
Gemäß Art 87 und 88 Medizinprodukteverordnung (VO-EU 2017/745) sind schwerwiegende Vorkommnisse und Gesundheitsbeschwerden von Anwendern von Medizinprodukten den zuständigen Behörden zu melden.
6. Welche Geräte-Arten –nach Art und Zahl aufgeschlüsselt (CPAP, BiPAP usw.) wurden bisher als defekt gemeldet?
7. Wie lange wurden die jeweiligen Geräte verwendet?
8. Wieviele Meldungen über schadhafte Beatmungs-Geräte seitens der Firma Habel/Philips sind beim BASG eingegangen
9. Wieviele Meldungen haben Privatpersonen beim BASG gemacht?
10. Wieviele Geräte und welche Gerätetypen davon hatten einen ersichtlich defekten Schaumstoff?
11.
Welche
gesundheitlichen Probleme insbesondere schwerwiegende[3] wurden
berichtet – sowohl bei der Firmenmeldung-als auch bei den
Privatpersonenmeldungen.
Die Gesundheitsprobleme bitte aufgeschlüsselt nach Bronchien/Lungen
betreffend sowie andere Gesundheitsprobleme.
Laut der früheren 11728/AB XXVII. GP – Anfragebeantwortung gibt es monatliche Status Updates des Herstellers und eine Abstimmung mit anderen Mitgliedstaaten des europäischen Wirtschaftsraumes. Laut aktueller Medizinprodukte-VO 2017/ 745, Art 43 sind Patienten zeitnahe und umfassend zu informieren.
12. Wann und wie werden betroffene und geschädigte Patienten über die Ergebnisse der Updates und die Zusammenarbeit im europäischen Raum informiert?
UNTERSTÜTZUNG von Patienten
Viele Patienten berichten in unterschiedlichen Medien vom Desinteresse öffentlicher Stellen und davon, mit dem Problem allein gelassen zu sein, obwohl sie kein Verschulden trifft. Sie werden lediglich auf den möglichen Rechtsweg verwiesen. Damit wird ein Herstellerproblem trotz aufrechter CE-Zertifizierung auf die Schultern einzelner, noch dazu geschädigter Patienten abgewälzt.
Die Patienten werden bzw. wurden mit ihrer Wahl zwischen „Pest und Cholera“ auch von den Fachgesellschaften im Stich gelassen, denn welche Entscheidung auch immer sie treffen/getroffen haben, ist sie mit einem hohen gesundheitlichen Risiko verbunden.
13. Wie werden die Patienten begleitet/betreut, wenn sie Gesundheitsprobleme berichtet haben – gibt es aufsuchende Hilfe - Unterstützungsangebote? Wenn ja, welche?
KOSTEN
Welche Schritte unternimmt das BMSGPK, im Hinblick auf die Krankenversicherungsträger, die die Kosten für diese Therapieform tragen. Die Kostenübernahme für diese Geräte setzt ja ein einwandfreies Funktionieren einer bestimmten Beatmungstherapie voraus. Nun ist es unbestritten, dass zahlreiche CE-zertifizierte Philips-Geräte nachweislich NICHT der vorgeschriebenen Qualität entsprechen/entsprochen haben.
14. Werden die Krankenversicherungen als Solidargemeinschaft der Versicherten zumindest die Kosten für die schadhaften Geräte vom Hersteller zurückverlangen?
Ein Kosten-Beispiel veranschaulicht die Zahlen:
pro Monat fällt eine Leihgebühr von ca. 140.-
Euro an, das sind pro Jahr 1680.-, für 5 Jahre 8400.- für 10 Jahre
sind das 16800 Euro nur Miete. Der Neuwert eines Geräts beträgt
einige Hundert bis etwa 3000-.-Euro je nach Type, Ausstattung und Zubehör.
Zu den Mietkosten kommen nun Kosten die für zahlreiche Untersuchungen
für die Abklärung der unspezifischen Symptomatik (ärztliche und Lungenfachärztliche
Konsultationen und Untersuchungen, CT, Lungenfunktion, Thorax-Röntgen, Sputum
Untersuchungen, Einstellung in Schlaflabor auf neues Gerät usw.)
Die vorsichtige Schätzung der Kosten die durch schadhafte Geräte
verursacht werden, ergibt pro Patient zumindest einige zigtausend Euro,
vermutlich zwischen (30-50.000.-€). Diese finanziellen Leistungen werden derzeit
statt vom Verursacher von der Solidargemeinschaft der Krankenversicherten
erbracht.
15. Wird diese
Vorgangsweise der Krankenversicherungen vom BMG hinterfragt?
Falls nein, warum nicht?
FACH-INFORMATIONEN an Ärztinnen und
Ärzte
16. Wie werden Ärzte und Lungenfachärzte von Amts wegen- da es keine private, sondern eine Angelegenheit des öffentlichen Gesundheitswesens ist - über mögliche Ergebnisse von wissenschaftlichen Untersuchungen zum Thema, Behandlungsmöglichkeiten informiert?
17. Werden sie über die aktuellen Berichte der FDA informiert?
18. Hat das BMK ein Netzwerk zum Informationsaustausch auf europäischer Ebene aufgebaut?
ANALYSE
19. Welche Maßnahmen hat das BASG der Firma Philips Respironics auferlegt, um die Ursachen für die Schaumstoff-Zersetzung zu analysieren und zu beheben?
20. Gibt es einen Auftrag zur Analyse der defekten Schaumstoffe? Wenn ja, wo wird untersucht? Ist diese Institution/en von Philips unabhängig?
UNABHÄNGIGE INFORMATIONEN[4]
21. Welche Maßnahmen setzt das BMSGPK, um solide Informationen über die Problematik zu bereit zu stellen, die unabhängig und unbeeinflusst von Aussagen der Fa. Philips Respironics sind? [5]
Diese Unabhängigkeit erscheint gerade in diesem Zusammenhang, in dem Philips, ein weltweiter börsennotierter Konzern, der nach wie vor auf seiner Website Gesundheitsprobleme negiert[6], andererseits eine dringliche Warnung über schadhafte Produkte herausgeben musste und die maßgebliche amerikanische Gesundheitsbehörde FDA nach wie vor von massenhaft auftretenden Gesundheitsproblemen und MDR-Meldungen berichtet? *(siehe Einleitung)
VERHINDERUNG von FOLGESCHÄDEN
Das Sicherheitsdatenblatt gibt Aufschluss über Zusammensetzung und Eigenschaften und mögliche toxikologische Wirkungen des Problematischen Schaumstoffs. Das Sicherheitsdatenblatt für diesen Schaumstoff (PE-PUR) ist eine zentrale Information, die eine Gefahreneinschätzung von Gesundheitsproblemen überhaupt erst ermöglicht. Die darin enthaltenen Detailinformationen sind Voraussetzung, um Gesundheitsprobleme beurteilen und eine medizinische Gefahreneinschätzung vornehmen zu können, entsprechende Untersuchungen oder Überwachung von krankhaften Veränderungen durchzuführen und mögliche Therapien einzuleiten. Ohne die Informationen des Sicherheitsdatenblatts sind die Ärzte der betroffenen Patienten im Blindflug unterwegs und können auch nicht helfen/therapieren, um gravierendere Schäden u.U. zu verhindern. Durch die aktive Verweigerung der Herausgabe dieser wesentlichen Information wird Patienten, die ohnehin durch den zerbröselten Schaumstoff/toxische Substanzen geschädigt sind, zusätzlich Schaden zugefügt.
Auch die dadurch entstehenden Folgekosten werden sich zusätzlich im Gesundheitssystem niederschlagen.
22. Wird das BMSGPK Maßnahmen setzen, damit die behandelnden Ärzte die Informationen des Sicherheitsdatenblatts erhalten, um eine sinnvolle zielgerichtete Arbeit für die Patienten zu ermöglichen?
23. Welchen Wert hat das verpflichtend zu erstellende Sicherheitsdatenblatt, wenn es im Problemfall, für den es gemacht wurde, geheim gehalten werden kann?
PATIENTENRECHTE
Die betroffenen Patienten sind in einer wirklich prekären Lage: sie wurden unwissentlich geschädigt, bekommen nun keine entsprechenden Informationen, um ihre gesundheitliche Schädigung angemessen erkennen und behandeln zu können und sind noch dazu mit dem juristischen Risiko gegenüber einem Industrie-Giganten, der alle Probleme abstreitet, allein gelassen.
24. Was beabsichtigt das BMSGPK, um den vielen Betroffenen, bereits geschädigten Patienten zu ihrem Recht auf zumindest angemessene DIAGNOSEN ihrer Gesundheitsprobleme zu verhelfen und damit eine zusätzliche Schädigung ihrer Gesundheit durch das Informationsdefizit zu verhindern?
25. Welche Maßnahmen ergreift das BMSGPK, damit die CE-Zertifizierung für Medizinprodukte immerhin handelt es sich hier um die Risikoklasse IIa - auch tatsächlich Sicherheit bedeutet und nicht nur ein sinnentleertes Symbol ist?
EPIDEMIOLOGISCHE AUFARBEITUNG[7]:
Da es etwa 35.000 Patientinnen und Patienten österreichweit betrifft, wird die Frage gestellt:
26. Wird das BMSGPK eine Darstellung der oben genannten Daten und Meldungen in Form eines epidemiologischen Reports das BASG beauftragen, wie das in anderen EU-Ländern geplant ist? Ähnlich wie in vergleichbaren epidemiologisch bedeutsamen Fällen.
27. Wird das BMSGPK eine unabhängige wissenschaftliche interdisziplinäre Aufarbeitung der Gesundheitsprobleme von Patientinnen und Patienten sicherstellen?
Es handelt sich aufgrund des Alters und der Häufigkeit von anderen Erkrankungen um eine vulnerable Gruppe, die auf Unterstützung seitens der öst. Behörden vertraut.
Es geht darum, künftig das Risiko für schwerwiegende Vorkommnisse zu verringern und von öffentlicher Hand sicher zu stellen. (lt. Medizinprodukte-VO 2017/745)